PAC-832: A New Drug for Alzheimer’s Disease

PAC-832 is the first drug of its kind — it works by selectively blocking a receptor called “galanin receptor 1,” or GalR1. The drug has sub-micromolar potency for GalR1 and >30x selectivity over GalR2/3. When administered to mice, it significantly improves their memory across multiple different memory tests. PAC-832 also has excellent manufacturability, low toxicity, and great pharmacokinetics, including passing the blood-brain barrier — all critical things needed for a neurological drug to succeed.
Motivation behind PAC-832
The primary symptom of Alzheimer’s disease (AD) — memory loss — has long been hypothesized to arise from the loss of neurons that produce the neurotransmitter acetylcholine (ACh) in certain important brain regions, primarily the basal forebrain. This is known as the “cholinergic hypothesis of AD,” which forms the basis for how the AD drug donepezil works. Donepezil increases the amount of ACh between neurons by blocking an enzyme called acetylcholinesterase (which is responsible for breaking down ACh). In doing so, it’s been shown to reverse memory loss in AD patients.
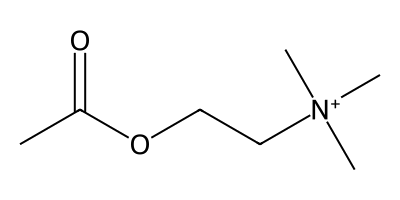
Donepezil is far from perfect and was discovered way back in 1983, yet shockingly the entire AD drug development field has been unable to improve on it after 40+ years of effort. During this time, the AD field has mostly focused on developing drugs that target amyloid-beta plaques and other “disease-modifying” mechanisms. This effort produced several new FDA-approved drugs in recent years which reduce plaque levels (like lecanemab and donanemab), yet none of these drugs meaningfully slow AD disease progression or outperform donepezil for managing AD symptoms, and they carry significant new safety risks like increased incidence of brain hemorrhage/stroke. Because of this, doctors are unwilling to prescribe these new drugs. Almost nobody uses them. Meanwhile, donepezil has remained the most effective, safe, and widely-used AD drug from its discovery up to today.
Given the underwhelming performance of anti-amyloid drugs, it’s a good time to focus on alternative AD drug mechanisms. I believe that the optimal place to look is the cholinergic pathway, which unlike more contemporary AD targets has a large body of the best possible supporting evidence — decades of real, human clinical data from cholinesterase inhibitors.
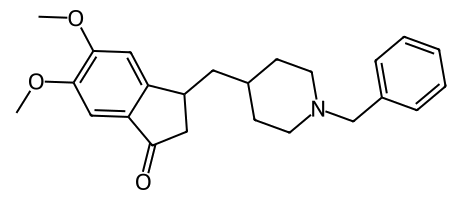
This line of thinking led me to develop PAC-832. Like donepezil and related cholinesterase inhibitors (galantamine, rivastigmine), PAC-832 ultimately increases ACh levels in the brain. However, PAC-832 is not structurally related to any existing cholinesterase inhibitors, and it doesn’t directly target the cholinergic pathway. Instead, it works through a new mechanism revolving around this complex, underappreciated molecule called “galanin.”
A brief history of the galanin-AD hypothesis
Galanin is a 30-amino acid neuropeptide — a small protein that acts as a signaling molecule in the brain. It was discovered in 1978. Though less well known than dopamine or serotonin, galanin is widely produced by neurons throughout the central and peripheral nervous system. Galanin signaling has been shown to regulate pain, metabolism, mood, sleep, and memory. It lurks in the background, quietly influencing many of our physiological functions on a daily basis.
Galanin dysregulation was first tied to AD pathology in the 1980s, 10 years after galanin’s discovery. Chan-Palay, Beal et al., and others observed that the basal forebrain from deceased AD patients contained many more galanin-producing neurons and higher galanin expression than healthy brains.


Meanwhile, Fisone et al. and Dutar et al. observed that galanin treatment onto slices from rat and monkey brains prevented the neurons from releasing ACh.


At the behavioral level, impaired memory was observed by McDonald et al. in rodents with galanin injected into their brains. Steiner et al. observed similar results in rodents genetically engineered to overexpress galanin.


Along with other supporting studies, these observations outlined a mechanism where galanin contributed to memory loss in AD by preventing ACh release in the basal forebrain, though neither galanin’s molecular mechanism nor the cause of galanin overexpression was understood at the time. This in turn led people in the 90s to hypothesize that a drug that ‘blocked’ the action of galanin could increase ACh levels and improve cognitive function in AD patients, similar to how cholinesterase inhibitors like donepezil work.

In the 2000s, the galanin-AD hypothesis was complicated by the emergence of data showing that galanin had neuroprotective effects in certain contexts. O’Meara et al. showed that galanin knock-out mice had significantly fewer cholinergic neurons in the basal forebrain, demonstrating that galanin was necessary for normal neuronal development. Meanwhile, Ding et al. observed that galanin protected cholinergic neurons from death caused by amyloid beta plaques. These findings, along with related reports, provided a surprising explanation for why cholinergic neurons in AD patients contained so much galanin in the first place — it was likely the brain trying to protect the neurons from neurotoxic factors brought on by AD progression, and the suppression of ACh release was collateral damage. This in turn suggested that a blanket inhibitor of galanin across all contexts ran the risk of causing harm by blocking galanin’s neuroprotective effects.


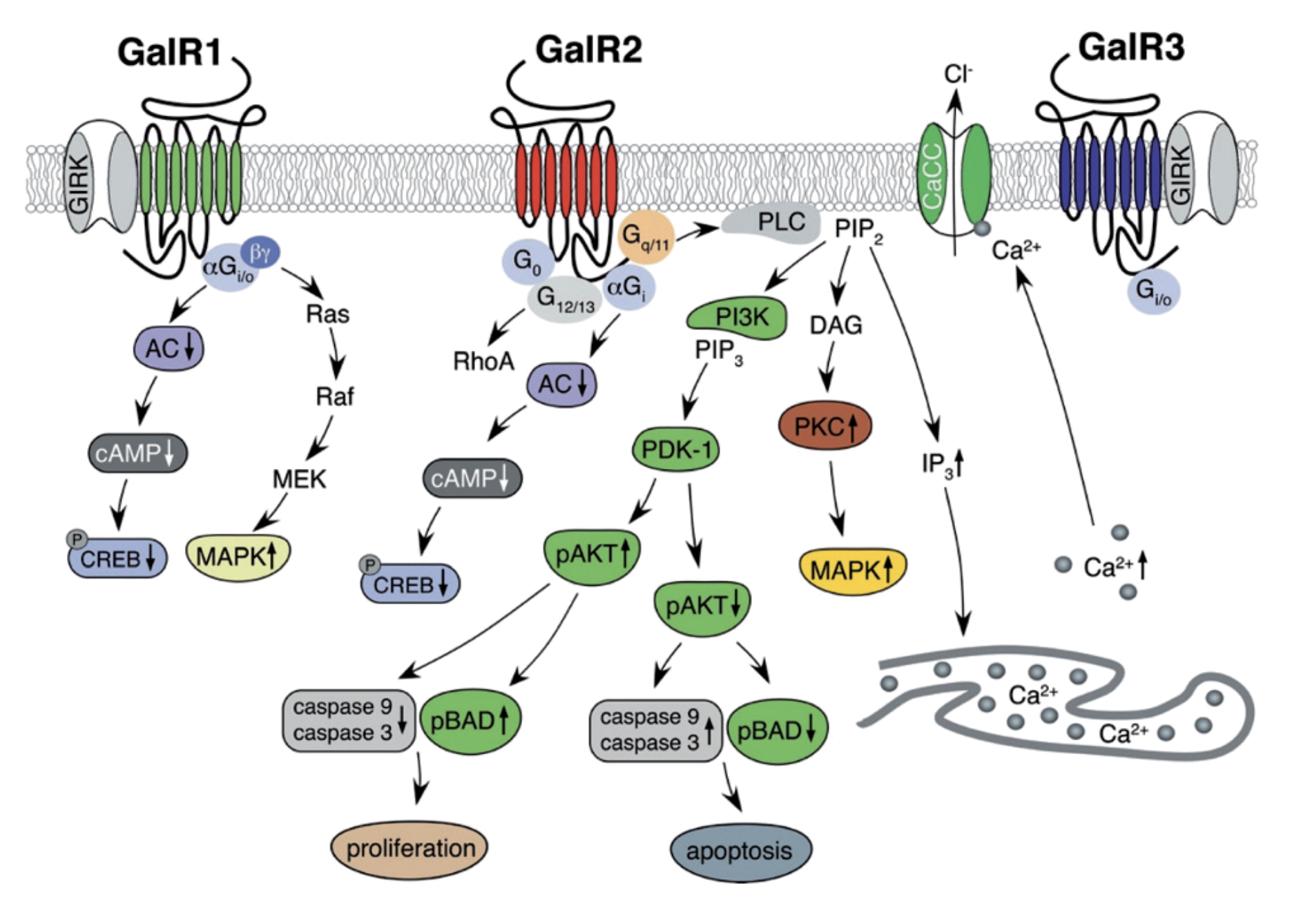
Concurrent molecular research helped resolve the seemingly contradictory neuroinhibitory vs neuroprotective effects of galanin. By the 2000s, it was known that galanin activated three different receptors, which were named galanin receptor 1, 2, and 3 (GalR1–3 for short). GalR1 and GalR3 were found to activate the “Gi/o signaling pathway,” a powerful molecular brake that suppresses neurotransmitter release. This signaling is desirable in certain non-AD contexts — for example, in overactive neurons prone to causing seizures. However, in cholinergic neurons relevant for memory, decreased ACh release is detrimental. This finding explains how galanin overexpression leads to ACh suppression and memory loss on a molecular level — via activating GalR1/3 and the Gi/o pathway.
Source: Lang et al. 2007, Pharmacology & Therapeutics
On the other hand, GalR2 was found to activate a different pathway — the Gq/11 pathway, which broadly results in cell activation; the opposite of Gi/o signaling. Moreover, various studies (Mahoney et al., Elliot-Hunt et al., and others) found that the neuroprotective effects of galanin were downstream of this pathway and mediated through GalR2.


A viable path for a pharmacological inhibitor of galanin emerged from these findings. Rather than blocking galanin activity across all contexts (which would also block its protective effects), one could in theory design an inhibitor that selectively blocks ONLY GalR1 and/or GalR3 (thus removing the brake on ACh release), while having no effect on GalR2 (thus preserving the protective Gq/11 signaling). Between GalR1 and GalR3, GalR1 was the more attractive target due to its higher expression in the brain.

Previous galanin antagonist drug development
At the same time that researchers were working out galanin’s biology in the 90s and onward, a parallel effort was underway to develop drugs that modulate galanin activity, with the hopes of finding a treatment/cure for any of the diseases tied to galanin dysregulation.
The initial batch of GalR antagonists/agonists developed in the 90s were all peptides. They were neither receptor subtype-specific nor able to pass the blood-brain barrier due to their size. They had to be administered by direct injection into the brain — feasible for a mouse, obviously not feasible for a human. Still, these early drugs provided important proof of concept data for galanin antagonist drugs. For example, McDonald and Crawley found that the peptide antagonist M40 (a galanin analog) was able to reverse cognitive deficits caused by galanin in rats.

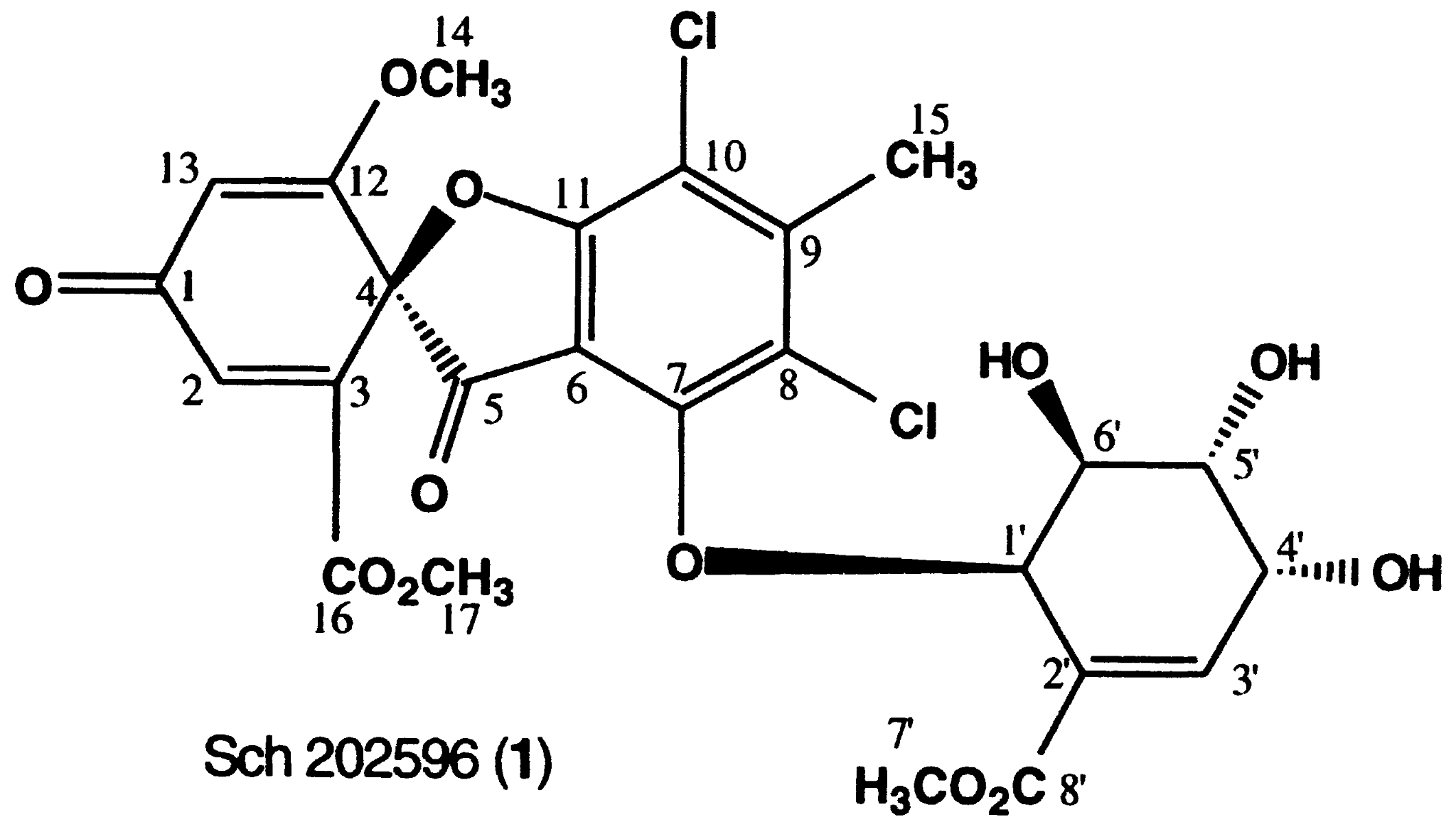
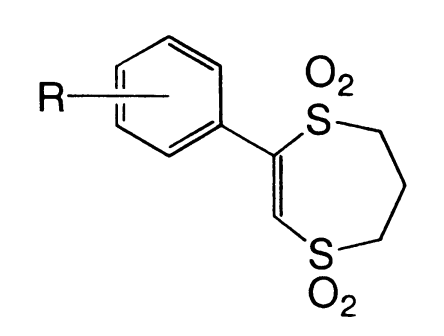
The first small molecule inhibitors of GalR1 with potential to pass the blood-brain barrier appeared in the early 2000s. A few high-throughput screens were carried out for GalR1 binding, producing two promising lead compounds/series with good affinity for GalR1 (King 1997, Scott et al. 2000). However, none of these compounds were viable for further development. One of them, Sch 202596, was a fungal metabolite that was too difficult to synthesize and modify. The other, the dithiin/dithiepine-1,1,4,4-tetroxide series, suffered from intractable “reactivity and solubility issues.”
Since then, there has been no further development of small molecule GalR1 antagonists — partially due to the practical challenges like the ones listed, and partially due to the fact that the galanin-AD hypothesis fell out of fashion in favor of other AD mechanisms. As of today, there are still no FDA-approved drugs that target galanin signaling. The closest thing so far has been the GalR3-selective small molecule antagonist HT-2157, which entered clinical trials in 2011 for depression. However, the drug did not progress beyond Phase 1 due to safety issues.
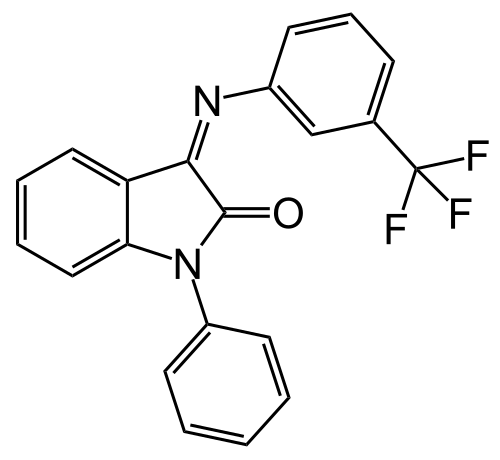
Enter PAC-832
To summarize everything so far: there is a large body of basic research dating back to the 1980s that supports galanin signaling as a promising target for treating cognitive decline in AD. However, various issues have prevented the development of a successful galanin antagonist drug, including (1) messy galanin biology that took decades to elucidate, which requires subtype-specific receptor antagonism that avoids GalR2, (2) poor chemical properties for the few small molecule GalR1 antagonists that were actually discovered, and (3) a lack of precedent from existing successful galanin-targeting drugs in the clinic.
PAC-832 solves issues (1) and (2), and will hopefully be the first drug to solve (3).

PAC-832 selectively inhibits only GalR1 and not GalR2/3. It has sub-micromolar potency toward GalR1 (IC50 = 0.28 μM) in an in vitro GalR1 activity assay, while showing no detectable activity at GalR2 or GalR3 at concentrations up to 10 μM. Thus, PAC-832 fits the desired profile of a galanin antagonist — potent inhibition of GalR1 to release the brake on ACh release, while having no effect on GalR2 to preserve neuroprotective signaling pathways.


The expected downstream effects of GalR1 inhibition are (1) increased ACh release, and (2) improved cognition. We confirmed that PAC-832 produces each of these observations in separate experiments. In an in vitro ACh release assay, PAC-832 concentration-dependently rescues ACh levels when co-administered with galanin, confirming (1).


Meanwhile, PAC-832 dose-dependently improves memory in two rodent behavioral assays, with comparable efficacy to donepezil, confirming (2).



Last but certainly not least, PAC-832 exhibits excellent developability. It has good stability, solubility, and ease of synthesis. It also has great pharmacokinetics, with 90% oral bioavailability and a brain-to-plasma ratio (Kp) of 0.7, as well as low toxicity.
I’m now preparing PAC-832 for Phase 1 clinical trials, which should be started within a year. PAC-832 will be the first GalR1 antagonist to reach the clinic.